
Obavijest o obvezi prijave izmjena za lijekove koji sadržavaju djelatne tvari: kandesartan, irbesartan, losartan, olmesartan i valsartan, nastavno na Odluku Europske komisije od 2. travnja 2019. vezano uz moguća onečišćenja N-nitrozaminima
06.05.2019.
Europska komisija je 2. travnja 2019. godine donijela Provedbenu odluku Komisije o odobrenjima za stavljanje u promet lijekova za humanu uporabu, koji sadržavaju djelatne tvari kandesartan, irbesartan, losartan, olmesartan i valsartan, u okviru članka 31. Direktive 2001/83/EZ Europskog parlamenta i Vijeća. Slijedom navedene Odluke države članice su obvezne izdati nacionalna odobrenja za stavljanje u promet lijekova koji sadržavaju djelatne tvari kandesartan, irbesartan, losartan, olmesartan i valsartan pod uvjetima utvrđenima u Prilogu II. i to temeljem znanstvenih zaključaka utvrđenih u Prilogu I. predmetne Odluke.
Za lijekove koji su u postupku odobravanja, uvjeti koji proizlaze iz odluke Komisije bit će implementirani u Rješenje o davanju odobrenja za stavljanje lijeka u promet s rokom ispunjenja najkasnije do 2. travnja 2021. godine.
Za već odobrene lijekove, u skladu s objavljenim preporukama na stranicama Koordinacijske grupe za postupak međusobnog priznavanja i decentralizirani postupak za humane lijekove (CMDh), nositelji odobrenja za stavljanje lijeka u promet obvezni su nacionalnim regulatornim tijelima:
- prijaviti izmjenu IAIN C.I.11.a u roku od 10 dana od objave Odluke Komisije radi implementacije uvjeta navedenih u Prilogu II. Odluke u odobrenje za stavljanje lijeka u promet. Temeljem prijavljene izmjene HALMED će nositelju odobrenja izdati Rješenje o izmjeni rješenja s implementiranim uvjetima i propisanim rokom za njihovo ispunjenje.
- prijaviti odgovarajuće izmjene u roku od 30 dana od objave Odluke Komisije radi ispunjavanja niže navedenih uvjeta koji moraju biti ispunjeni u vrijeme donošenja Odluke:
a) Nositelj odobrenja za stavljanje lijeka u promet mora osigurati da je za serije djelatnih tvari koje se upotrebljavaju u njegovim lijekovima uspostavljena kontrolna strategija za sve N-nitrozamine.
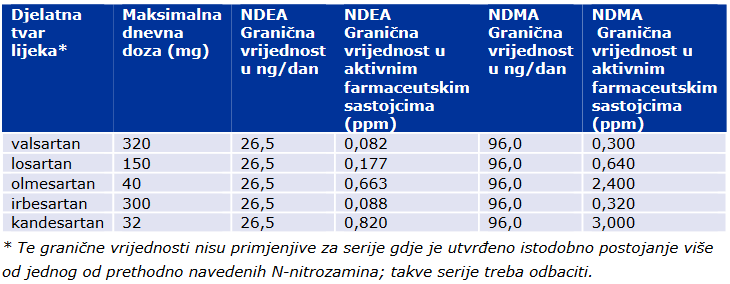
b) Za N-nitrozodimetilamin (NDMA) i N-nitrozodietilamin (NDEA) nositelj odobrenja za stavljanje lijeka u promet mora uvesti sljedeće specifikacije za djelatnu tvar lijeka:
U prijelaznom razdoblju od dvije godine potrebno je primjenjivati granične vrijednosti za tvari NDMA i NDEA navedene u nastavku:
Detalji o izmjenama koje je potrebno prijaviti u svrhu ispunjenja uvjeta i njihova klasifikacija dostupni su u obavijesti o zaključcima sa sjednice CMDh-a iz ožujka 2019. godine. U skladu sa zaključkom CMDh-a iz travnja 2019. godine, izmjene koje se tiču implementacije uvjeta moguće je i grupirati.




