
Notice of obligation to submit variations for medicinal products containing active substances candesartan, irbesartan, losartan, olmesartan and valsartan, further to the April 2, 2019 European Commision Decision regarding possible N-nitrosamines impuritie
07.05.2019
On April 2, 2019 the European Commission adopted a Commission Implementing Decision concerning the marketing authorisations of medicinal products for human use which contain the active substances candesartan, irbesartan, losartan, olmesartan and valsartan, in the framework of Article 31 of Directive 2001/83/EC of the European Parliament and of the Council. Following that Decision, the Member States concerned shall subject the national marketing authorisations for the medicinal products which contain the active substances candesartan, irbesartan, losartan, olmesartan, valsartan to the conditions identified in Annex II on the basis of the scientific conclusions set out in Annex I.
For currently ongoing marketing authorisation application procedures, the conditions stemming from the Commission's Decision will be implemented in the marketing authorisation with the deadline for fulfillment no later than April 2, 2021.
For existing marketing authorisations, in accordance with the published recommendations by the Co-ordination Group for Mutual Recognition and Decentralised Procedures - Human (CMDh), marketing authorisation holders have to:
- Submit a type IAIN C.I.11.a variation within 10 days after publication of the Commission Decision, for inclusion of the conditions on the review and change of the manufacturing process and the final limits for NDMA and NDEA in the specifications for the active substance into the marketing authorisation. Based on the submitted variation, HALMED will issue the Amendment of Marketing Authorisation to the MAH with the deadline for the fulfillment of the implemented conditions.
- Submit appropriate variations within 30 days after publication of the Commission Decision, in order to fulfill the following conditions that must be met at the time of the Decision:
a) For all N-nitrosamines the MAH must ensure a control strategy is in place in drug substance batches used for their drug products
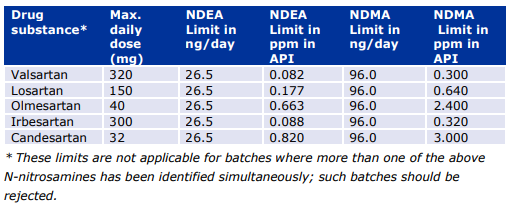
b) For N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) the MAH must introduce the following specifications for the drug substance:
Limits for NDMA and NDEA outlined below should be implemented for a transitional period of 2 years
Details on the variations that need to be submitted in order to fulfill the conditions, as well as their classification, are available in the report from the CMDh meeting held in March 2019. In accordance with the report from the CMDh meeting held in April 2019, variations regarding the implementations of conditions can be grouped, as appropriate.




